Vinylový kation
Vinylový kation je karbokation s kladným nábojem na alkenovém uhlíku; s empirickým vzorcem C2H +
3 . Obecněji může jít o jakýkoliv disubstitovaný trojvazný atom uhlíku, v němž nabitý atom uhlíku je součástí dvojné vazby a má hybridizaci sp. V chemické literatuře sem bývá zařazována širší skupina, než jsou jen ionty obsahující C2H +
3 .
Vinylové kationty jsou jedním z hlavních druhů reaktivních meziproduktů a mají rozmanitou reaktivitu. Vyskytují se při solvolytických reakcích[1][2] a elektrofilních adicích na alkyny,[3] kde se utvářejí protonacemi alkynů pomocí silných kyselin. Jako i jiné sp hybridizované sloučeniny mají vinylové kationty převážně lineární tvar.
K příbuzným sloučeninám patří allylové, benzylové a arylové karbokationty.

Na rozdíl od jiných reaktivních meziproduktů, jako jsou radikály a karboanionty byly vinylové kationty po dlouhou dobu málo prozkoumané[4] a původně se předpokládalo, že mají příliš vysoké energie, než aby mohly být meziprodukty reakcí. Vinylové kationty byly jako reaktivní meziprodukty poprvé navrženy v roce 1944 pro kysele katalyzovanou hydrolýzu alkoxyacetylenů na alkylacetáty.[5]
V prvním kroku, který určoval rychlost reakce, se měl vytvářet kation s kladným nábojem na dikoordinovaném uhlíku, jednalo se o první popsaný případ takového meziproduktu.
Historie
editovatDetekce vinylových kationtů se podařila až 15 let po jejich zahrnutí do reakčního mechanismu; nalezeny byly u solvolytických reakcí alfa-vinylhalogenidů.[6][7]
Výzkum vinylových kationtů pokračoval v 60. letech, kdy byla získána kinetická data podporující jejich existenci; například D. Noyce popsal vytváření vinylových kationtů při kysele katalyzované hydrataci kyseliny fenylporopiolové.[8] V kroku určujícím rychlost vznikl kladný náboj na benzylovém atomu uhlíku, což ukazovalo na průběh reakce přes vinylkation. K vysvětlení jeho tvorby posloužily hyperkonjugace a vodíkové vazby.
Vznik
editovat
Vinylové kationty se objevují v solvolytických reakcích, majících jako SN1 reakce kinetiku prvního řádu. Vinylhalogenidy bývají v roztocích nereaktivní: dusičnan stříbrný nesráží za jejich přítomnosti stříbrné halogenidy,[10] což bylo používáno jako argument proti existenci vinylkationtů.[4] Zavedení „super“ odstupujících skupin v 70. letech umožnilo tvorbu vinylových kationtů s dostatečnou životností.[11]
Tyto skupiny, jako jsou trifláty (trifluormethansulfonáty) a nonafláty (nonafluorbutansulfonáty), snadno vstupují do SN1 reakcí. Jejich využití umožnilo potvrdit existenci vinylkationtů.

Byly nalezeny i další odstupující skupiny, například jodany, což jsou millionkrát lepší odstupující skupiny než běžné trifláty.[13]
R. J. Hinkle se svými spolupracovníky připravil řadu alkenyl(aryl)jodoniových triflátů z hypervalentních fenyljodových prekurzorů. Na následujícím obrázku je vyobrazena tvorba E- a Z-vinyltriflátů heterolytickým štěpením vazeb uhlík-jod a následným zachycením kationtu triflátem. Přítomnost E- i Z- vinyltriflátových produktů podporuje předpoklad o vytváření primárních vinylových kationtů; pokud by reakce probíhala SN2 mechanismem, tak by vznikal jen jeden izomer.[9]
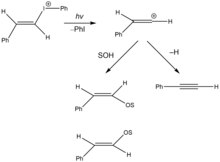
Vinylové kationty byly rovněž vytvořeny fotochemickými solvolýzami. Na obrázku vpravo je znázorněna fotochemická solvolýza vinyljodoniové soli heterolýzou vazby uhlík-jod, za vzniku vinylkarbokationtu a jodbenzenu.[14] Reaktivní meziprodukt se může účastnit nukleofilního ataku rozpouštědla za tvorby E- a Z-izomerů enoletheru, nebo beta-hydridové eliminace.
Vznik cyklických vinylových kationtů
editovatSnadnost vytváření cyklických vinylových kationtů závisí na velikosti kruhu, menší kruhy vznikají obtížněji, což je v souladu s výpočty ukazujícími, že vinylové kationty upřednostňují lineární uspořádání atomů uhlíku.[15]
Vzhledem k vysokému kruhovému napětí u 3členných kruhů nebyl nejmenší teoreticky možný vinylový kation, cycloprop-1-enyl, dosud zachycen.[16] SN1 solvolýza používaná na přípravu ostatních vinylových kationtů se u cykloprop-1-enylového iontu ukázal jako neúčinný.
Struktura
editovat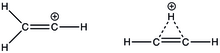
3 [17]

Nejjednodušší vinylový kation, C2H +
3 , má dvě možné struktury, lineární a můstkovou. Můstková je oproti lineární stabilnější, a to o 21 kJ/mol.[17] U substituovaných vinylových iontů obsahujících alkylové skupiny je, jak bylo zjištěno pomocí 13C a 1H NMR.[18] Prvními experimentální důkazy lineární struktury vinylových kationtů byly získány rentgenovou krystalografickou analýzou β-silylvinylových iontů. Ve vícejaderné NMR spektroskopii se objevil jeden signál 29Si NMR, což naznačuje, že oba atomy křemíku jsou rovnocenné, protože jsou po karbokationtu delokalizované prostřednictvím hyperkonjugace. Vinylový kation v infračerveném spektru vykazuje silnou absorpci na 1987 cm−1, odpovídající natahování vazby C=C+. Vazebné úhly mezi uhlíkovými atomy vinylového iontu a prvními uhlíky alkylových substituentů jsou blízké 180°.[19]
Stabilita
editovat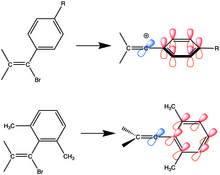
Existence vinylových kationtů byla sporná z důvodů jejich velkých rozdílů energie oproti vinylovým prekurzorům. Jakmile byly získány stálé vinylové kationty solvolýzou vinylových sloučenin s dobrými odstupujícími skupinami, například trifláty a nonafláty, stabilizované skupinami dodávajícími elektrony, tak byl dosažen v této oblasti významný pokrok.
Jeden z prvních zkoumaných vinylových kationtů měl arylové substituenty obsahující skupiny dodávajícími elektrony. Arylvinylové sloučeniny jsou stabilizované rezonancí. Po oddělení odstupující skupiny je prázdný orbital p kolmý ke konjufovanému systému fenylového jádra, takže jeho přechodný stav dosáhne rezonanční stabilizace pouze tehdy, když je prázdný orbital p u vinylu ve stejné rovině jako p systém fenylového jádra. Přidáním sterických efektů na pozice ortho zlepší konjugaci, protože se fenylový kruh stane kolmým k vinylovým uhlíkům, ale nachází se v rovině prázdného orbitalu p.



Podobně jako arylvinylkationty jsou i odpovídající dienylové a alenylové ionty stabilizovány konjugací. I zde musí být dvojné vazby ve stejné rovině jako prázdný orbital p, aby mohlo dojít ke stabilizaci. U allenylových kationtů je kladný náboj rozmístěný po celé struktuře.
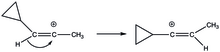
Cyklopropylvinylové ionty jsou stabilizovány méně obvyklým způsobem. V jejich rozpůlené struktuře je dostatečný překryv prázdných p orbitalů a cyklopropylového kruhu. U kolmé struktury jsou prázdné p orbitaly ke kruhu kolmé. Stabilizace vyvolávaná cyklopropyly je zde natolik velkou termodynamickou řídící silou, že při solvolýze (E)- a (Z)-3-cyklopropyl-2-propenyltriflátů vyvolává 1,2-hydridové přesmyky.[20]
Vliv substituentů na stabilitu vinylových kationtů
| Substituent | Stabilizace# | Elektronové efekty α-substituentu | ||
| Indukce^ | π-donace | Hyperkonjugace | ||
| -CH˭CH2 | + | - | +* | |
| -CH3 | + | + | ||
| -Cl | + | - | +* | |
| -Br | + | - | +* | |
| -I | + | - | +* | |
| -F | - | -* | + | |
| -NH2 | + | - | +* | |
| -OH | + | - | +* | |
| -SH | + | - | +* | |
| -C6H5 | + | +* | ||
| -CF3 | - | - | ||
| -CH2F | - | - | ||
| -NO2 | - | - | ||
| -C≡N | + | - | +* | |
| -CH2Y*** | + | - | +* | |
| -Si(CH3)3 | + | + | ||
| -C(O)H | - | +/-** | ||
| -COOH | - | +/-** | ||
| -C(CH3)2OH | + | - | ||
| -C≡CH | + | - | +* | |
^ ‘-’ odtahuje elektrony, ‘+’ dodává elektrony
# + označuje stabilizaci a - destabilizaci substituovaného vinylového kationtu oproti odpovídajícímu neutrálnímu alkenu
*označuje jev, který má největší vliv na stabilizaci nebo destabilizaci u substituentů vytvářejících více než jeden elektronový efekt
** znamená, že substituent je indukčně oddělen od karbonylového uhlíku a delokalizace elektronů není výrazná
*** Y = -F, -Cl, -Br, -I, -OH, -CN, -CF3
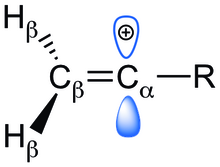
Přítomnost prázdných p orbitalů kolmých na vazby π způsobuje destabilizaci vinylových iontů. Tuto nestabilitu lze zmírnit vhodnými interakcemi s α-substituenty, které zmenšují náboj karbokationtu. Ke zkoumání stabilizačních a destabilizačních vlivů substituentů bylo použito pozorování změn entalpií, délek a řádů vazeb a struktur, společně s ab initio výpočty.

Existují tři druhy elektronových efektů, kterými substituenty mohou ovlivňovat stabilitu vinylových kationtů. Může jít o destabilizaci dalším snížením elektronové hustoty na uhlíku nebo stabilizaci jejím zvýšením. Kladný náboj karbokationtu se může zmenšit působením nenasycených uhlíkatých nebo heteroatomových substituentů π-donací a/nebo hyperkonjugací vazeb C-H methylenovými/methylovými skupinami. Indukční efekty mohou mít stabilizační i destabilizační účinky, závisí na tom, jestli substituenty dodávají nebo odtahují elektrony. Jednotlivé elektronové efekty od sebe nelze oddělit, protože celkovou stabilitu kationtu ovlivňují všechny.

Stabilitu vinylových kationtů lze srovnat s jejich neutrálními alkenovými analogy. Aby byly zjištěny stabilizační účinky α-substituentů, tak byly použity isodesmické reakce na výpočty rozdílů entalpií mezi substituovanými vinylkationty a jejich neutrálními alkenovými prekurzory jako entalpie těchto reakcí.
Na začátku byly zkoumány 4 substituenty (-CH=CH2, -F, -Cl, -CH3) a vliv jejich elektronových efektů na stabilitu vinylových kationtů. Různé α-substituenty vytvářely změny struktury kationtů oproti příslušným neutrálním alkenům; tyto změny lze připsat elektronovým efektům. Významná zkrácení vazeb C-R a C=C by naznačovala dodávání elektronů nebo indukci mezi Ca a R a Cb a Ca. Případný nárůst délky vazby Cb-H by znamenal silný vliv hyperkonjugace a termodynamickou stabilizaci iontu. Stabilizaci umožňuje dobrý překryv vazeb C-H s prázdnými p orbitaly vazeb Ca. Hyperkonjugace je přítomná u všech struktur.
Výpočty entalpií na základě isodesmických reakcí byly v souladu s experimentálními daty. Míra stabilizace se zvyšuje v tomto pořadí: -F < -Cl < -CH3 < -CH=CH2. Stabilizační účinky měly všechny substituenty kromě fluoridového, který ionty destabilizoval o přibližně 30 kJ/mol. Destabilizační účinky fluoru lze vysvětlit efekty, kterými α-fluorový substituent působí na vinylové a ethylové kationty. Ethylové kationty fluor stabilizuje. Rozdíl ve vlivu fluoru na stabilitu vinylového a ethylového kationtu je způsoben rozdílnými hybridizacemi α-uhlíků. Vinylový kation má sp-hybridizovaný uhlík, jenž je elektronegativnější a tím roste účinek indukčních efektů. Interakce elektronegativních sp-hybridizovaných atomů uhlíku s atomy fluoru strukturu destabilizují. Obdobný jev se v menší míře vyskytuje mezi –CH3 an–CH=CH2, kde -CH=CH2 stabilizuje méně.
Heteroatomy jako fluor a chlor mohou vyvolávat jak indukční efekty (odtahující elektrony), tak i π-donaci, kdy dodávají elektrony, protože mají vysoké hodnoty elektronegativity a rovněž π-elektrony. Stabilizace pak závisí na vzájemném působení těchto jevů. U fluoru převažuje indukční destabilizace a rezonance je výrazně slabší. V případě chloru je rezonance silnější než indukce a ve výsledku tak dochází ke stabilizaci.
Při navázání indukčně odtahujících/dodávajících a π-dodávajících substituentů zůstávají částečné náboje na R a Cα. Dochází také k nárůstu řádu vazby mezi Cβ=Cα a Cα-R, což odpovídá změnám v jejich délkách.
U malých substituentů nebyla pozorována žádná souvislost mezi řádem vazby a změnou rozdělení náboje na R a stabilizací vyvolanou substituentem. Stabilizace ovšem souvisela s prodlužováním vazeb Cβ-H.
Na základě výše uvedených mechanismů lze široké rozmezí α-substituentů vinylových kationtů rozdělit podle jejich elektronových efektů, na jejichž vzájemném vyvážení závisí míra stabilizace.
Substituenty s volnými elektronovými páry, jako například –NH2, -OH a –SH vinylové kationty stabilizují, protože zde π-donace převažuje nad indukčními efekty. Konjugované systémy, jako –CH=CH2 a –C6H5, stabilizují v důsledku silné π-donaci. Silně destabilizující substituenty, kam mimo jiné patří –CF3 a –NO2, vykazují pouze indukční odtahování elektronů. Slabě destabilizující substituenty, například –CN, mají slabou π-donaci, která nedokáže překonat odtahování elektronů.
Nelze plně oddělit indukční efekty heteroatomových α-substituentů, protože se objevují i jiné elektronové efekty. Indukční efekty ovšem lze zkoumat pomocí efektů vytvářených β-substituenty, kde heteroatomy mohou být od nabitého uhlíku odděleny methylenovou skupinou (-CH2Y). U –CH2Y s malou nebo žádnou π-donací jsou jen malé rozdíly mezi vlivy hyperkonjugace –CH2- skupin substituentů. Celkovou stabilitu tak převážně ovlivňují efekty β-substituentů, řízené pouze indukcí. Indukční efekty vyvolávané navázanými skupinami klesají v řadě: CN > CF3 > F > Cl > Br > OH, přičemž některé destabilizační energie jsou podobné jako u methylové skupiny.
Ve většině případů vytvářejí substituenty více než jeden stabilizující či destabilizující efekt. Indukční efekty tvořené vícenásobnými vazbami na heteroatomy jsou často překonávány π-donací stejného heteroatomu, například čistě podle β-indukce je -CN silnější než CF3, ale v důsledku pπ-donace z dusíkového atomu v CN je jeho indukce snížena. U obvyklých heteroatomových substituentů, jako jsou F, Cl, Br a OH, se stabilizace snižuje s rostoucí schopností odtahovat elektrony; přesto se stále projevuje π-donace, což je vidět na zkrácení vazby C-R.
Karbonylové substituenty bývají převážně destabilizující kvůli silnému částečnému kladnému náboji na karbonylovém uhlíku vedle vinylového kationtu a nepřítomnosti π-donace.
Působení substituentů na vinylové a ethylové kationty je vhodným prostředkem ke zkoumání stabilizačních efektů hybridizace. Vinylové kationty obecně bývají substituenty stabilizovány více než ethylové kationty, hlavně proto, že samotné vinylové kationty jsou méně stabilní. U skupin se silným indukčním odtahováním elektronů, což jsou například –F, -OH a –NH2, je indukční destabilizace výraznější na vinylových než na ethylových kationtech, jelikož vysoce elektronegativní povaha vinylových kationtů, jež jsou sp-hybridizované, oproti sp2-hybridizovaným ethylovým iontům. Oproti tomu α-Si(CH3)3 více stabilizuje vinylové kationty, protože neobsahuje π-elektrony.
Stabilizující substituenty zvyšují řády vazeb C-R, Cα=Cβ a Cβ-H. Malé nárůsty řádů vazeb byly pozorovány u –CF3, -CH2F a –CH2X, které nejsou schopné π-donace, zatímco větší změny se objevují u substituentů schopných dodávat π-elektrony, tedy CH=CH2, -I a –SH.[21][22]
Vinylové kationty jako meziprodukty chemických reakcí
editovatElektrofilní adice
editovat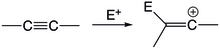
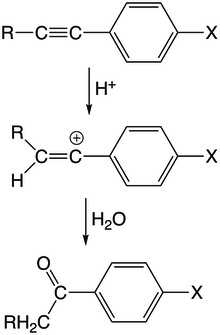
Vinylkationtové meziprodukty se objevují u reakcí, kde elektrofilní částice atakují nenasycené uhlovodíky. Kladně nabitý elektrofil zde reaguje s jedním z nenasycených atomů uhlíku a vytvoří vinylový kation, který následně projde dalšími reakcemi a vytvoří se konečný produkt.
V kysele katalyzovaných hydratacích arylacetylenů proton nejprve atakuje trojnou vazbu za vzniku vinylového kationtu s nábojem na arylovaném uhlíku. Tento meziprodukt se vyznačuje mírnou rezonanční stabilizací, protože konjugovaný arylový orbital je kolmý na prázdný orbital π vinylového kationtu. Reakce je prvního řádu vzhledem k acetylenu i protonu a protonace acetylenu je krokem určujícím rychlost reakce. Monosubstituované aryl/alkoxyacetyleny v kysele katalyzovaných hydratacích reagují rychleji než jejich methylované protějšky. U arylacetylenů je stabilizační vliv methylových skupin menší, než jaký vytvářejí atomy vodíku, protože se objevuje hyperkonjugace vazeb C-H; tento stabilizační trend je tak opačný než u alkylkationtů. Tato hyperkonjugace má velký význam, protože vazby C-H mívají výrazné překryvy s prázdnými π-orbitaly. Možným vysvětlením této stabilizace je také to, že vodíkové substituenty se v důsledku své menší velikosti snadněji solvatují.
Acetyleny mohou být atakovány i jinými elektrofily než protony. V případě karboxylových kyselin mohou vznikat cis/trans alkeny. Reakcemi s halogenovodíky, při nichž také dochází k protonacím, vedou ke vzniku halogenovaných alkenů. Adamantylketony mohou vznikat ataky adamantanylových kationtů na acetylen a následnou hydratací.[24]
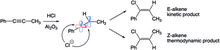
Při hydrohalogenaci fenylpropenu vznikají v důsledku termodynamických a kinetických efektů dva různé alkeny. Lineární sp-hybridizovaný kation může být atakován halogenem ve dvou různých směrech. Při ataku ze stericky méně zatížené strany (na vodíku) se vytvoří E-alken, v opačném případě vzniká Z-alkenový produkt. V krátkém časovém měřítku převládá E-izomer, protože atak z méně stíněné strany probíhá rychleji, po delší době ovšem převládne stabilnější Z-alken (s methylovými a fenylovými skupinami na opačných stranách). E-alken, jenž se tvoří ze začátku, se izomerizuje na Z-alken přes karbokation vzniklý protonací a rotací vazby C-C.[25]


Skupiny sousedící s násobnou vazbou mohou urychlit reakce prostřednictvím vnitromolekulárních interakcí s meziprodukty. Alkyn sousedící s terciární alkoholovou skupinou vytváří jako meziprodukt čtyřčlenný cyklický vinylový kation, ve kterém kyslíkový atom hydroxylové skupiny dvojicí vazeb propojuje dva atomy uhlíku. Podobně vzniká pětičlenný chloroniový kruh z 5-chlor-pent-1-ynu. Vzniká přitom neobvykle posunutý produkt, což má za příčinu heterolýzu v poloze C5-Cl.[24]

V elektrofilních adicích na alleny probíhají elektrofilní ataky způsobem upřednostňujícím vytváření koncových aduktů a vinylových kationtů s nábojem na centrálním uhlíku. Polarizace allenové skupiny naznačuje, že koncové uhlíky mají vyšší elektronovou hustotu a jsou náchylné k nukleofilním atakům. Pokud je ovšem koncový uhlík stabilizován vhodným substituentem, tak mohou vznikat elektrofilními ataky na centrálním uhlíku allylové ionty. Podobně jako u fenylových kruhů sousedících s vinylovými kationty je k dosažení plné rezonanční stabilizace potřebná rotace vazby.[24]
Přesmyky u vinylových kationtů
editovat
Vinylkationtové meziprodukty vzniklé v průběhu reakcí často podléhají přesmykům. Tyto přesmyky lze rozdělit do dvou skupin: přesuny na dvojné vazby a přes dvojné vazby. Do první patří 1,2-přesmyky vedoucí k tvorbě allylových kationtů, zatímco u druhého typu se vytváří jiný izomer vinylového kationtu.
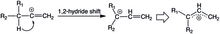
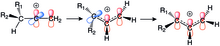
Vinylové kationty podstupují 1,2-hydridové přesmyky na allylové (stabilizované) kationty. Tento druh přesmyku je u alkylových kationtů běžný a z hlediska NMR probíhá rychle; u vinylových kationtů je ovšem vzácný, přestože jsou produkty takových přesmyků termodynamicky stálé. Podobně jako u arylovaných vinylkationtů jsou orbitaly interagující při přeměně lineárních vinylových kationtů na nelineární allyly navzájem kolmé a reakce probíhají přes nerovinné meziprodukty, kvůli čemuž jsou přesmyky obtížně proveditelné; to se projevuje vyššímiaktivačními energiemi 1,2-hydridových přesmyků vinylových kationtů oproti kationtům alkylovým. Jako příklady reakcí, které byly pozorovány, lze uvést protonace dialkylovaných alkynů a solvolýzu ispropylvinyltrifluormethansulfonátu v trifluorethanolu.
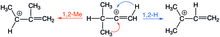


U vinylových kationtů mohou probíhat také 1,2-methylové přesmyky; podobně jako u 1,2-hydridových mají vyšší aktivační energie než v případě odpovídajících alkylových iontů. Při protonacích alkynů se mohou objevit 1,2-hydridové i 1,2-methylové přesmyky, který z nich bude převažovat, závisí na alkylovém substituentu, jenž určuje výsledný allylkationtový produkt. U terc-butylových substituentů převažují methylové přesmyky a u isopropylových přesmyky hydridové. Cyklické alkeny po solvolýze podstupují 1,2-methylové přesmyky.

U solvolýz spiro-vinyltriflátů vznikají vinylkationtové meziprodukty soustředěnými procesy vyvolávajícími další přesmyky a výrazné změny cyklických struktur. Přesmyky vinylových kationtů je dokonce možné využít k rozšiřování kruhů.

Druhou skupinu přesmyků tvoří ty, kde se vinylové kationty mění na jiné, izomerní. Průběh těchto procesů záleží na použitém rozpouštědle, vlastnostech nukleofilu a funkčních skupinách navázaných na ion. U primárních vinylových kationtů jsou 1,2-hydridové přesmyky vzácné, protože tyto ionty jsou málo stabilní v důsledku slabé schopnosti vodíku dodávat elektrony. Mohou se ovšem objevit v některých vzácných případech jako je 1-methyl-2-fenylvinyltriflát, kde je příslušný vinylový kation rezonančně stabilizován.


Methylový přesmyk probíhá také, když se terc-butylový kation přeměňuje na but-2-yn. Pentaallylový kation zde vzniká jedním 1,3-methylovým přesmykem nebo dvěma po sobě jdoucími 1,2-přesmyky. Přesmyky přes dvojné vazby mohou i měnit velikosti cyklů. Při solvolýze methylovaného cyklohexenyltriflátu vznikají přesmyknutý a nepřesmyknutý produkt v téměř stejných množstvích, s jen malou převahou produktu přesmyku. Produkt methylencyklopentanového přesmyku ovšem vykazuje určité pnutí.
I halogeny se mohou přesunout a stabilizovat tak vinylové kationty. Při reakci 5-chlorpentynu s kyselinou trifluoroctovou dochází současně k protonaci a 1,4-ku chloru, který vytváří můstkovou cyklickou strukturu přes čtyři atomy uhlíku. Kyselina trifluoroctová následně atakuje meziprodukt za vzniku 2-chlorpent-4-enyltrifluoracetátu; podobná reaktivita se vyskytuje i u ostatních halogenů, například fluoralkyny vytvářejí produkty se dvěma adukty.[18]
Vinylové kationty v pericyklických reakcích
editovat
Keteny a alleny vstupují do tepelných [2+2] cykloadicí. Tyto adice probíhají soustředěným mechanismem, protože π orbitaly substrátů jsou navzájem kolmé. Vinylkationtové meziprodukty procházejí stejným procesem, a to z toho důvodu, že obsahují dva p orbitaly, které se mohou současně překrýt s orbitaly dienofilu. V Smirnovově-Zamkowově reaguje but-2-yn s chlorem a vzniká dichlorcyklobutan. Podobně reaguje allen s kyselinou chlorovodíkovou; cykloadicí se vytváří cyklický kationtový meziprodukt, jenž je poté atakován nukleofilem za vzniku konečného produktu.[26]
Reference
editovatV tomto článku byl použit překlad textu z článku Vinyl cation na anglické Wikipedii.
- ↑ T. Okuyama. Solvolysis of Vinyl Iodonium Salts. New Insights into Vinyl Cation Intermediates. Accounts of Chemical Research. 2002, s. 12. DOI 10.1021/ar0100374.
- ↑ R. Gronheid. Thermal and Photochemical Solvolysis of (E)- and (Z)-2-Phenyl-1-Propenyl(phenyl)iodonium Tetrafluoroborate: Benzenium and Primary Vinylic Cation Intermediates. Journal of the American Chemical Society. 2001, s. 8760. DOI 10.1021/ja010861n.
- ↑ Andrew J. Walkinshaw; Wenshu Xu; Marcos G. Suero; Matthew J. Gaunt. Copper-Catalyzed Carboarylation of Alkynes via Vinyl Cations. Journal of the American Chemical Society. 2013, s. 12532-12535. DOI 10.1021/ja405972h. PMID 23947578.
- ↑ a b P. J. Stang. Vinyl Cations. New York: Academic Press, 1979. S. 2.
- ↑ Thomas L. Jacobs; Scott Searles. Acetylenic Ethers. IV.1 Hydration. Journal of the American Chemical Society. 1944-05-01, s. 686–689. ISSN 0002-7863. DOI 10.1021/ja01233a007.
- ↑ C. A. Grob. Die Solvoltische Decarboxylierung von α,β-Ungesättigeten β-Halogensäuren Fragmentierungsreaktionen, 9. Miteilung. Helvetica Chimica Acta. 1964, s. 1590. DOI 10.1002/hlca.19640470621.
- ↑ K. Miyamoto. Facile Generation of a Strained Cyclic Vinyl Cation by Thermal Solvolysis of Cyclopent-1-Enyl-λ3-Bromanes. Angewandte Chemie International Edition. 2009, s. 8931–8934. DOI 10.1002/anie.200903368. PMID 19830754.
- ↑ D. Noyce. Concerning the Acid-Catalyzed Hydration of Acetylenes. Journal of the American Chemical Society. 1965, s. 2295. DOI 10.1021/ja01088a042.
- ↑ a b R. J. Hinkle. Primary Vinyl Cations in Solution: Kinetics and Products of a,a-Disubstituted Alkenyl(aryl)iodonium Triflate Fragmentations. Journal of the American Chemical Society. 1999, s. 7437–7438. DOI 10.1021/ja9916310.
- ↑ R. L. Shriner. Systematic Identification of Organic Compounds. New York: Wiley, 1964. Dostupné online.
- ↑ Michael Hanack. Vinyl cations in solvolysis reactions. Accounts of Chemical Research. 1970-07-01, s. 209–216. ISSN 0001-4842. DOI 10.1021/ar50031a001.
- ↑ P. J. Stang. Vinyl Cations. New York: Academic Press, 1979.
- ↑ Tadashi Okuyama; Tomoki Takino; Takuya Sueda; Masahito Ochiai. Solvolysis of Cyclohexenyliodonium Salt, a New Precursor for the Vinyl Cation: Remarkable Nucleofugality of the Phenyliodonio Group and Evidence for Internal Return from an Intimate Ion-Molecule Pair. Journal of the American Chemical Society. 1995-03-01, s. 3360–3367. ISSN 0002-7863. DOI 10.1021/ja00117a006.
- ↑ a b Thomas T. Tidwell; J. P. Richard. Advances in physical organic chemistry. Vol. 37. [s.l.]: [s.n.], 2003-01-01. Dostupné online. ISBN 978-0120335374.
- ↑ Herbert Mayr; Reinhard Schneider; Dieter Wilhelm; Paul V. R. Schleyer. Vinyl cations. Comparison of gas-phase thermodynamic and solvolysis data with ab initio MO calculations. The Journal of Organic Chemistry. 1981-12-01, s. 5336–5340. Dostupné online. ISSN 0022-3263. DOI 10.1021/jo00339a015.
- ↑ C. A. Grob; J. Csapilla; G. Cseh. Die solvoltische Decarboxylierung von α,β-ungesättigeten β-Halogensäuren Fragmentierungsreaktionen, 9. Miteilung. Helvetica Chimica Acta. 1964, s. 1590–1602. ISSN 1522-2675. DOI 10.1002/hlca.19640470621.
- ↑ a b c J. A. Pople. The structure of the vinyl cation. Chemical Physics Letters. 1987, s. 10–12. DOI 10.1016/0009-2614(87)80294-4. Bibcode 1987CPL...137...10P.
- ↑ a b c d e f g h i j k l A. A. Shchegolev; M. I. Kanishchev. Rearrangements in Vinyl Cations. Russian Chemical Reviews. 1981, s. 553–564. DOI 10.1070/rc1981v050n06abeh002650. Bibcode 1981RuCRv..50..553S.
- ↑ Thomas Müller; Mark Juhasz; Christopher A. Reed. The X-ray Structure of a Vinyl Cation. Angewandte Chemie International Edition. 2004-03-12, s. 1543–1546. Dostupné online. ISSN 1521-3773. DOI 10.1002/anie.200352986. PMID 15022228.
- ↑ a b c d e Michael Hanack. Stabilized vinyl cations. Accounts of Chemical Research. 1976-10-01, s. 364–371. ISSN 0001-4842. DOI 10.1021/ar50106a004.
- ↑ Kaj van Alem; Gerrit Lodder; Han Zuilhof. α-Substituted Vinyl Cations: Stabilities and Electronic Properties. The Journal of Physical Chemistry A. 2000-03-01, s. 2780–2787. ISSN 1089-5639. DOI 10.1021/jp9935743. Bibcode 2000JPCA..104.2780V.
- ↑ Kaj van Alem; Gerrit Lodder; Han Zuilhof. Delocalization Does Not Always Stabilize: A Quantum Chemical Analysis of α-Substituent Effects on 54 Alkyl and Vinyl Cations. The Journal of Physical Chemistry A. 2002-11-01, s. 10681–10690. ISSN 1089-5639. DOI 10.1021/jp021766j. Bibcode 2002JPCA..10610681V.
- ↑ a b c Advances in Physical Organic Chemistry. archive.org. Academic Press, 1971-12-31, s. 185. Dostupné online. ISBN 9780080581484.
- ↑ a b c Giorgio Modena. Vinyl cations. Advances in Physical Organic Chemistry. 1971, s. 185–280.
- ↑ a b Organic Chemistry. [s.l.]: Oxford University Press Dostupné online. ISBN 9780199270293.
- ↑ a b Ian Fleming. Molecular Orbitals and Organic Chemical Reactions, Reference Edition - Fleming - Wiley Online Library. [s.l.]: [s.n.], 2010. ISBN 9780470689493. DOI 10.1002/9780470689493.
Externí odkazy
editovat Obrázky, zvuky či videa k tématu vinylový kation na Wikimedia Commons
Obrázky, zvuky či videa k tématu vinylový kation na Wikimedia Commons